

Sandwich ELISA Protocol

1. Introduction to ELISAs
The Enzyme-Linked Immunosorbent Assay (ELISA) is a powerful antibody-based technique for detecting and measuring a specific analyte, such as a protein, within a complex liquid sample. It can be used qualitatively to simply confirm if a target is present, or quantitatively to determine its exact concentration. A key advantage of ELISA over methods like a Western blot (WB) or immunohistochemistry (IHC) is its ability to provide precise quantitative data for a large number of samples efficiently. It is commonly used to measure biomarkers, hormones, and proteins like cytokines and chemokines.
2. Key Decisions
2.1 Types of ELISA
There are four main types of ELISA: Direct, Indirect, Sandwich and Competitive.
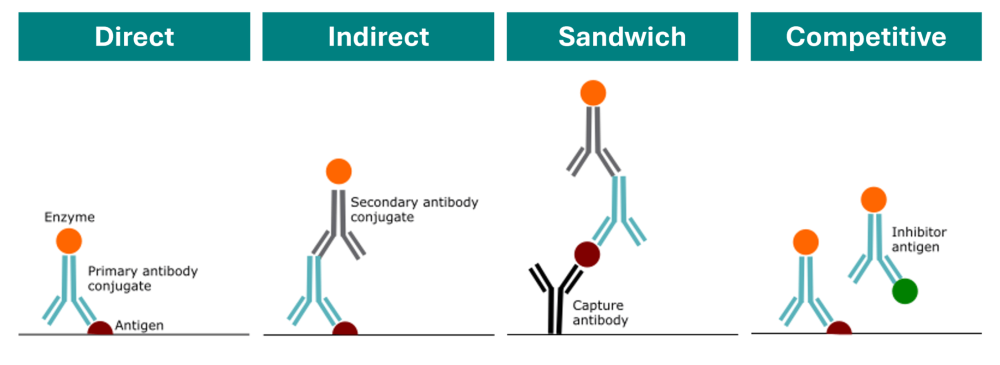
2.1.1 Direct
- In a direct ELISA, the antigen is immobilized in the well of an ELISA plate. The antigen is then detected by an antibody directly conjugated to an enzyme such as HRP.
- Due to the fewer steps required, direct ELISAs are much faster than other ELISA techniques and less prone to error due to fewer reagents being used. However, the disadvantages of direct detection methods include higher background noise in comparison to indirect ELISA due to non-specific immobilization of the antigen. Direct ELISA is also less flexible due to the requirement of a specific conjugated primary antibody needed for each target protein. Furthermore, as no secondary antibody is used, there is no signal amplification and therefore reduced assay sensitivity.
2.1.2 Indirect
- The indirect ELISA method involves a two-step detection method, where the antigen is immobilized onto the ELISA plate and labeled with an unlabeled primary antibody which binds the specific antigen. Then an enzyme conjugated secondary antibody directed against the host species of the primary antibody is applied.
- The indirect ELISA method has a higher sensitivity compared to direct methods due to the use of more than one labeled secondary antibody that can bind the primary antibody. It also provides greater flexibility as different primary antibodies can be used with a single labeled secondary antibody. Disadvantages of indirect ELISAs include the possibility of cross-reactivity of secondary antibody to the adsorbed antigen which may increase background noise. Indirect ELISAs also require longer protocols due to the additional incubation time of the secondary antibody required.
2.1.3 Sandwich
- A sandwich ELISA features the use of matched antibody pairs, known as capture and detection antibodies, which recognize the same target analyte but bind to different epitopes. Using the sandwich analogy, the antigen is the “filling” whilst the two antibodies are seen as the “bread” capturing the filling. The capture antibody is used to coat the ELISA plate and binds the antigen which can then be detected in a direct or indirect ELISA configuration.
- To visualize and quantify the reaction, the detection antibody is either labelled directly or a secondary step included in the assay. Addition of a soluble substrate is then used, and the resulting color is directly proportional to amount of analyte present in the sample solution.
- The key advantage of the sandwich ELISA is its high sensitivity in comparison to direct and indirect ELISAs. Sandwich ELISAs also have a high specificity due to the use of two antibodies to detect the antigen. The sandwich method also offers flexibility due to the ability to use either direct or indirect methods. The disadvantages of the sandwich method include time required to optimize the antibody pair to avoid cross-reactivity between capture-detection antibodies, if a standardized ELISA kit or tested antibody pair is not available.
2.1.4 Competitive
- A competition/inhibition ELISA, also known as a blocking ELISA, is the more complex of the ELISA techniques. The competitive ELISA is commonly used to measure the concentration of an antigen in a sample by detecting interference in an expected signal output. The sample antigen or antibody “competes” with a reference for binding to a limited amount of labeled antibody or antigen, respectively. The higher the sample antigen concentration, the weaker the output signal, so the signal output inversely correlated with amount of antigen in the sample.
2.2 Colorimetric vs Fluorescent ELISA
- Traditionally ELISAs use colorimetric systems for signal detection, where a colored compound is produced following catalysis of a substrate by an enzyme (e.g. HRP or AP). The benefit of using a colorimetric assay being able to use the relatively common and cheap plate readers that are widely available in most labs. The most widely used colorimetric ELISA substrate is TMB which is available in multiple strenths as either HB9992 - TMB ELISA Substrate (standard) or HB8566 - TMB ELISA Substrate (high sensitivity)
- Fluorescent ELISAs work by direct labelling of an antibody with a fluorophore excited by a specific wavelength of light, which then emits light at a slightly longer wavelength. Fluorescence offers the ability to carry out multiplex arrays (probing for multiple antigens at the same time).
- Chemiluminescence enables the detection of sub-picogram protein concentrations and relies on the emission of light as a product of enzyme-mediated catalysis of a substrate. A commonly used substrate is luminol, which in the presence of HRP and H2O2 is oxidized to 3-aminophthalate. Unlike colorimetric assays, light is only present whilst the reaction is occurring. Once the substrate is exhausted, the signal stops. The assay must be measured using a luminometer or a plate reader with luminescent capabilities. Hello Bio offers a choice of two high quality ECL substrate kits that are compatible with ELISA assays: HB7090 - SuperBlot™ ECL Western Blotting Substrate Kit (Standard) and HB9308 - SuperBlot™ ECL Western Blotting Substrate Kit (High sensitivity)
2.3 Controls
It is crucial to include the necessary controls when conducting any ELISA experiment to be confident of outcome validity.
- Negative control: Sample that does not contain analyte of interest i.e. contains only buffer.
- Plate blank: Used for subtraction from other wells when reading the plate. Typically, 3-8 wells per assay. Blanks should include TMB and stop solution and be blocked and washed as all the other wells, but no reagent or sample added.
- Other negative controls can be included as needed i.e. for cell culture supernatant you may wish to include cell culture medium alone.
- Samples should be analyzed at minimum in duplicate.
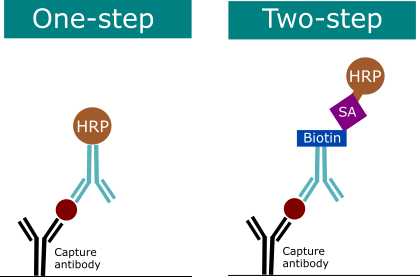
2.4 One or Two Step Detection
In an ELISA, either the detection antibody is directly labeled (one-step detection) or the detection antibody itself is detected in a second step (two-step detection). Two-step detection methods are considered more sensitive due to signal amplification.
- One-step detection uses a HRP conjugated detection antibody. Some applications may benefit from this due to the slightly reduced sensitivity due to high abundance of analyte of interest in the sample.
- Two-step detection uses a detection antibody conjugated with biotin, streptavidin is added which binds non-covalently to biotin molecules in a highly specific manner.

2.5 Choosing an enzyme and substrate pair
ELISAs are performed with colorimetric detection with selected ELISAs based on chemiluminescence or fluorescence. Chromogens are chemical compounds that are converted via a chemical reaction to produce a color. The use of these compounds in ELISA is popular due to the ability to see the result by eye and the requirement of commonly used photometers to perform readouts. The substrate chosen will depend on the type of enzyme used in the ELISA reaction:
- Horseradish peroxidase (HRP) is an enzyme found in the root of horseradish and catalyzes the colorimetric reaction of the assay. HRP is more commonly used due to its rapid kinetics. However, it should be noted that HRP is degraded in presence of antibacterial agents or sodium azide. When running an ELISA based on HRP, it is recommended to use TMB (3,3’,5,5’ tetramethylbenzidine). When added to the reaction, TMB produces a soluble blue product. TMB substrate is colorless in its reduced form and changes to blue once in the presence of HRP. Incubate for 15 minutes at room temperature away from direct light. The reaction is stopped by the addition of an acid (we recommend 0.16M sulfuric acid). The stopped reaction is then measured at 450nm. Hello Bio provides a range of products designed for HRP ELISAS:
- Alkaline peroxidase (AP) is a mammalian enzyme found in two forms: tissue and intestinal. The AP reaction is slower than HRP but has a more linear activity. Also, in contrast to HRP, AP activity is not affected by the presence of antibacterial agents or sodium azide. If running an ELISA based on AP, it is recommended to use pNPP (p-Nitrophenyl Phosphate). This reaction typically takes longer than HRP-TMB, and it is recommended to take multiple readings at 30, 60 and 90 minutes. The plate should be incubated at room temperature away from direct light. pNPP produces a soluble yellow product which is measured at 405nm.
3. Equipment and Consumables
|
Equipment |
Consumables |
|
|
4. Protocol
This protocol is designed for colorimetric two step detection using a biotinylated detection antibody and streptavidin HRP.4.1 Safety
Always follow local rules and read the full COSHH document for any chemical that you have not used previously. Always wear appropriate PPE such as a lab coat and gloves.
Specifically highlighted hazards:
- The Stop Solution is a strongly acidic solution. When diluting concentrated acids always add acid to water and not the other way around. Work in a fume hood. Wear appropriate PPE when handling this reagent.
4.2 Sandwich ELISA Protocol
- Coat a 96-well high-binding ELISA microplate with 100µl/well capture antibody – incubate overnight at 4°C.
- When coating the capture antibody onto the ELISA plate, recommended concentrations are 0.5-4µg/ml but this will need optimisation
- There are a range of different coating buffers that are used which differ in their effectiveness depending on what protein is being coated:
- Carbonate-bicarbonate buffer (0.1M, pH 9.6) is the most popular coating buffer. The higher pH helps solubility of many proteins and peptides and ensures most are protonated with an overall negative charge which aids binding to the positively charged plate.
- 0.1M carbonate buffer (pH8 - 8.2) is another effective coating buffer using less basic conditions than the standard carbonate-bicarbonate buffer.
- PBS / TBS are also widely used as they keep the protein at a physiological pH but are often less efficient at coating.
- It’s important to ensure the plate used facilitates the binding of high amounts of protein. For ELISAs it is recommended to use flat-bottomed plates usually made of polystyrene which enables the binding of proteins via hydrophobic bonding. Our protocol is designed for 96-well plates, with 100µl being sufficient per well for reagents and 200-300µl per well used for each wash step. While it is possible to coat plates at room temperature with a few hours incubation it is highly recommended to incubate overnight at 4°C.
- Block plate with 200µl/well blocking buffer (2% BSA in PBST) – incubate for 1 hour at RT.
- Blocking the plate ensures free binding sites in the well are saturated, eliminating the possibility of non-specific binding. Different protein blockers can be used, however bovine serum albumin (BSA) is the most popular and well documented choice for blocking.
- Wash 3 x times 300µl/well wash buffer (PBST or TBST).
- In the ELISA procedure, assay reagents are added in excess to ensure saturation. To avoid unspecific binding and carryover, it is important to sufficiently wash the plate between steps. This can be done manually or by using an automated washer. Wash buffers are composed of PBS and a detergent such as Tween-20 (0.02%). Volumes used in wash steps are recommended to be higher than those used when adding reagents as this ensures no residual reagent is bound to the walls of the wells. After washing excess buffer is removed by tapping the plate on absorbent paper. Be careful to move straight onto the next step, not allowing the plate to dry!
- Add 100µl/well sample or standard diluted in assay diluent. Incubate for 2 hours at RT or overnight at 4°C.
- All samples and standards should be diluted a minimum of 1:2 in the appropriate ELISA dilution buffer. The composition of buffer used with vary depending on sample type i.e. for cell culture supernatant it is recommended to use PBS with 0.05% Tween-20 and 0.1% BSA.
- Following reconstitution of the standard, prepare serial dilutions of the standard in the same buffer used for sample dilutions. Dilutions used should cover the standard range and be prepared no longer than 30 minutes before use.
- Wash 3 x times 300µl/well wash buffer (PBST or TBST).
- Add 100µl/well biotinylated detection antibody diluted to recommended concentration with blocking buffer. Incubate 1 hour at room temperature.
- If no recommended concentration is available this will need optimising with a good concentration range being 0.1 - 3µg/ml.
- Wash 3 x times in 300µl/well wash buffer (PBST or TBST).
- Add 100µl/well streptavidin HRP-conjugate. Incubate 30 mins at room temperature.
- This may need optimising depending on the ELISA with there being a wide range of dilutions commonly used from 1:1000 all the way to 1:50,000.
- Wash 3 x times in 300µl/well wash buffer (PBST or TBST).
- Develop with 100µl/well detection reagent (keep out of direct light).
- For TMB incubate for 15 minutes at room temperature before then stopping the reaction using 100µl/well stop buffer (0.16M H2SO4)
- For pNPP incubate for up to 90 minutes at room temperature and take multiple readings at 30, 60 and 90 minutes.
- Use a plate reader to measure absorbance.
- For TMB measure at 450nm
- For pNPP measure at 405nm
5. ELISA Data Analysis
The readout provided by the plate reader provides optical density (OD) values for each well. These can be used to compare samples of unknown concentrations to the known concentration of serially diluted standards. Firstly, subtract the OD of the reference wavelength measurement from each well (some plate reader software makes this subtraction for you if appropriate parameters set). The plate blank can also be subtracted from all values before analysis (again, certain plate reader software may do this for you as well).5.1 Quantitative or Qualitative?
- ELISA assays enable researchers to achieve a quantitative measure of protein abundance present in a sample. This is calculated by comparison of optical density (OD) values of a sample to that of a standard curve, whereby the sample protein concentration is interpolated.
- ELISAs can also be used to more simply provide qualitative data, indicating whether the antigen of interest is present in the sample or not. This is achieved by comparison of the sample to a negative control (blank) and a positive control expressing the antigen (ideally present at a low level to provide a lower threshold for antigen detection).
- ELISAs can also be “semi-quantitative” where the analyte concentration is compared to that of a positive control.
5.2 Standard Curves?
- The standard curve in an ELISA assay is used to determine the concentration of a chosen analyte in unknown samples. This is achieved through analysis of known amounts of purified protein serially diluted and analyzed alongside the samples of unknown concentration. The standard curve is generated by plotting the absorbance values of standard wells of known concentrations.
- The standard curve plot is generated by averaging the standard replicates for each concentration then subtracting the absorbance values for the blank control. These values are then plotted, and an appropriate curve fitted. The absorbance values of the unknown samples can then be compared to the standard curve to interpolate the protein concentration.
- A standard curve can simply be made by drawing straight lines between points of each concentration; however, this method will not provide an estimate between the standard data points and would not provide an accurate value of the true curve. Therefore, generally plotting a standard curve involves modeling of the standard data to generate a line equation which can be used to predict concentrations of the samples. How this model is fitted is very important to the accuracy of the results of an ELISA assay.
- The simplest model to fit is a linear regression, which uses the linear range of an assay. The goodness of fit, which is how well the model describes the data, is determined by the R2 value, where R2 value > 0.99 is considered a very good fit.
- To generate a standard curve, plot the log10 of the standard concentration against the log10 of the OD. Some plate reader software feature more advanced model fitting options that allow automatic generation of the standard curve, in this case it is recommended to choose a curve with a 4- or 5-parameter fit.
- When quantifying, don’t forget to adjust the determined sample concentrations by multiplying by the dilution factor. If the OD value of the sample falls outside of the standard range, it is advised to repeat the assay and dilute the sample at a higher dilution factor.
6. Reagents
- 0.1M Carbonate - Bicarbonate buffer pH9.6 - Add 5.76g/L NaHCO3 and 3.33g/L Na2CO3 then check that the pH is 9.6 and adjust if necessary.
- 0.1M Carbonate buffer pH8.2 - Add 8.4g/L NaHCO3 and check that the pH is 8-8.3
- 10x PBS pH7.4 - Add 80g/L NaCl, 2g/L KCl, 14.4g/L Na2HPO4, 2.4g/L KH2PO4 and adjust pH to 7.4. Alternatively PBS is available as HB5330 - PBS (100 Tablets).
- PBST (0.02% Tween-20) - Add 100ml/L 10x PBS then add 0.2ml/L Tween-20 (for accurate pipetting it is easier to make a 10% Tween-20 stock then add 2ml/L of 10% Tween-20). Alternatively PBST is available with 0.1% Tween-20 as HB8088 - PBS buffer with Tween 20 (20x)
- 10x TBS pH7.4 - Add 80g/L NaCl and 24.2g/L Tris base then adjust pH to 7.4. Alternatively TBS is available as HB7121 - TBS (25x) (pH 7.4)
- TBST (0.02% Tween-20) - Add 100ml/L 10x TBS then add 0.2ml/L Tween-20 (for accurate pipetting it is easier to make a 10% Tween-20 stock then add 2ml/L of 10% Tween-20). Alternatively TBST is available with 0.1% Tween-20 as HB6971 - TBS-T with Tween 20 (20x)
- Blocking Buffer (2% BSA in PBST / TBST) - Add 2g/100ml BSA to either TBST or PBST depending on what buffer the ELISA is being run in.
7. Troubleshooting
|
Problem |
Potential cause |
Suggested Solutions |
|
High background |
Insufficient washing |
Increase number of washes Add 30 second soak step in between washes |
|
Too much streptavidin-HRP |
Check dilution, titrate if needed |
|
|
Insufficient blocking |
Increase blocking time Check blocking solution dilution calculation |
|
|
Interfering substances in sample or standard |
Run appropriate controls
|
|
|
Incubation time too long |
Reduce incubation times |
|
|
Buffers contaminated |
Make up fresh buffers |
|
|
No Signal |
Reagents incorrectly prepared or added in incorrect order |
Repeat assay Check calculations, make up new buffers, standards etc Review protocol steps |
|
Contamination of HRP with azide |
Use fresh reagent |
|
|
Insufficient antibody used |
Increase antibody concentration |
|
|
Standard expired (see signal in sample wells) |
Check standard handled appropriately according to directions Use new vial of standard |
|
|
Capture antibody did not bind to plate |
Check correct plate used (must be ELISA-compatible plate, not a tissue culture plate) Dilute in PBS without additional protein |
|
|
Buffers contaminated |
Make fresh buffers |
|
|
Too much signal – plate uniformly blue |
Insufficient washing – unbound HRP remaining |
Increase number of washes Add 30 second soak step in between washes |
|
Incubation time is too long |
Follow recommended incubation times for each step |
|
|
Overly concentrated streptavidin-HRP used |
Check dilution, titrate is necessary |
|
|
Reused plate sealers or reagent reservoirs. Resulting in presence of residual HRP, resulting in non-specific color change of TMB solution. |
Use fresh plate sealer and reagent reservoir for each step. |
|
|
Contaminated buffer – residual HRP |
Make fresh buffers |
|
|
Standard curve has poor discrimination between points/low or flat curve |
Insufficient streptavidin-HRP |
Check dilution, titrate if necessary |
|
Capture antibody did not bind to plate |
Check correct plate used (must be ELISA-compatible plate, not a tissue culture plate) Dilute in PBS without additional protein |
|
|
Insufficient detection antibody |
Check dilution, titrate if necessary |
|
|
Plate not developed long enough |
Increase incubation time with substrate |
|
|
Procedure carried out incorrectly |
Check protocol, remove any modifications |
|
|
Errors in calculation of standard curve dilutions |
Check calculations, repeat standard curve with new dilutions |
|
|
Poor duplicates |
Insufficient washing |
Increase number of washes Add 30 second soak step in between washes |
|
Uneven plate coating |
Check correct plate used (must be ELISA-compatible plate, not a tissue culture plate) Dilute in PBS without additional protein Check coating and blocking volumes, times and method of reagent addition. Check plate used |
|
|
Reused plate sealer |
Use fresh plate sealer for each step |
|
|
No plate sealer used |
Use plate sealers |
|
|
Buffers contaminated |
Make fresh buffers |
|
|
Poor assay to assay reproducibility |
Insufficient washing |
Increase number of washes Add 30 second soak step in between washes |
|
Variations in incubation time/temperature |
Carry out incubations according to recommended time and temperatures Avoid incubating plates where environmental conditions may vary |
|
|
Variations in protocol |
Adhere to same protocol steps from assay to assay |
|
|
Plate sealer reused |
Use fresh plate sealer for each step |
|
|
Incorrect calculation of standard curve dilutions |
Check calculations, make new standard curve dilutions Use internal controls |
|
|
Buffers contaminated |
Make fresh buffers |
|
|
No signal in sample wells |
Sample matrix masking detection |
Dilute samples minimum 1:2 in sample diluent, or do series of dilutions to look at recovery |
|
Sample signal too high, standard curve looks fine |
Samples contain target analyte in levels above assay range |
Dilute samples and repeat assay |
|
Low readings across plate |
Incorrect wavelengths selected |
Check filters/plate reader parameters |
|
Insufficient development time |
Increase development time |
|
|
Coated plates used are too old |
Coat fresh plates |
|
|
Capture antibody did not bind to plate |
Check correct plate used (must be ELISA-compatible plate, not a tissue culture plate) Dilute in PBS without additional protein |
|
|
Green color develops upon addition of stop solution when using streptavidin-HRP |
Reagents not mixed sufficiently |
Tap/shake plate |
|
Edge effects |
Uneven temperatures around plate area |
Avoid incubating plates in areas where environmental conditions may vary Use plate sealers |
|
Drift |
Interrupted assay set-up |
Assay set-up should be continuous, have all reagents/standards prepared before commencing assay |
|
Reagents not at room temperature |
Ensure all reagents at room temperature before beginning assay |
8. Further Reading
- Lindstrom and Wager, 1978. IgG Autoantibody to Human Serum Albumin Studied by the ELISA-Technique. Scandanavian Journal of Immunology. doi: 10.1111/j.1365-3083.1978.tb00472.x
- Kato et al., 1977. Use of Rabbit Antibody IgG Bound onto Plain and Aminoalkylsilyl Glass Surface for the Enzyme-Linked Sandwich Immunoassay. Journal of Biochemistry. doi: 10.1093/oxfordjournals.jbchem.a131678
- Yorde et al., 1976. Competitive enzyme-liked immunoassay with use of soluble enzyme/antibody immune complexes for labeling. I. Measurement of human choriogonadotropin. Clin Chem. PMID: 949847
- Engvall and Perlmann., 1971. Enzyme-linked immunosorbent assay (ELISA) quantitative assay of immunoglobulin G. Immunochemistry. doi: 10.1016/0019-2791(71)90454-X